
Version-1.4 (aka 2004.0) Release Notes, February 2003
This release marks the first Staden Package release made available
since Rodger Staden's group disbanded at MRC (due to funding
issues). Since then MRC released the package under an Open Source
licence and the package has migrated to SourceForge.net.
One important change is the focus of development. The main body of
changes here have been implemented by myself (James Bonfield) at the
Wellcome Trust Sanger Institute, with the exception of the Contig
Navigation function (by Mark Griffiths, also of WTSI). It is clear
that ALL changes apply to Gap4 and Prefinish. This is as a direct
consequence of the group disbanding and my new position at WTSI. If
there are areas of the package that you feel are now being neglected
(which there certainly are!) and you are in a position to help, please
email me using 'jkbonfield at users.sourceforge.net'.
One other important change is the level of support
available. Basically no support is guaranteed, and no support should
be expected. However PLEASE do submit any bugs you find (even if it's
in part of the package no longer being actively worked on) to the
SourceForge bug tracking system. It may be found at:
https://sourceforge.net/tracker/?group_id=100316&atid=627058
Currently the package has been built for most Unix systems we
previously supported (possibly all - check the SourceForge downloads
links to check), however MS Windows support is likely to be lagging
behind. This is primarily because I do not yet have the resources
available to maintain a set of MS Windows binaries. Again, if you can
assist in this area please contact me.
Now, on to the changes themselves. The full list is rather long and
only included below incase you are a glutton for punishment. A
summaried version follows.
Gap4 Tag Editing
This has had quite a major overhaul. The most obvious change is that
the Edit Tag and Delete Tag functions have now become cascading menus
showing all the tags under the current editing cursor position. This
solves the problem of how to manipulate tags that are not the top-most
displayed ones, but it does mean editing is slower.
Therefore to speed up editing the F11 and F12 keys are bound to edit (F11)
and delete (F12) the top-most visible tag. Note that you will need to
enable F12 in the Edit Modes menu as by default it is disabled.
Tag Macros are another new feature. Using Shift-F1 to Shift-F10 a tag
editor window will appear. From here you can set the tag type, strand
and comment and then use "Save Macro" to remember those settings. They
are remembered for this session only, so use the editor Settings ->
Save Macros to store these to disk. Once defined, a tag macro may be
applied by underlining the region you wish to tag and then pressing
the appropriate function key, F1 to F10.
By positioning the editor cursor above a tag and using Control-F1 to
Control-F10 you can take a copy of the tag underneath the cursor and
store it in the appropriate tag macro (without bringing up the macro
editor). Combined with F1 to F10 this then provides an equivalent to
tag cut-and-paste.
The Tag Editor window itself has also undergone some changes. The
existing Save command works just as before, but there are now two new
ways to Save; Move and Copy. To use these firstly underline a new
region that you wish to tag. Move then moves the tag to that region
while Copy creates a duplicate at that region. The underlined region
does not need to be within the same sequence, or even the same contig
editor. (Although it does need to be an editor within the same Gap4!)
Contig Navigation
At the editor level right clicking on a sequence name now shows a
submenu "goto...". This contains a list of the readings sharing that
template. The sub-menu can be torn off if you wish.
Hyperlinks have also been added to the reading list produced in the
editor using Settings -> Set Active Readings. Just double click on a
name in the list to goto that reading in the contig editor. Similarly
hyperlinks are now on lists loaded in via the main Lists menu.
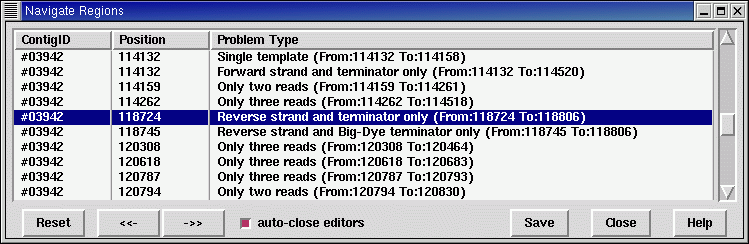
However by far the most flexible way of navigating through contigs
using an external file is the Contig Navigation function (in main gap4
View menu). This needs a filename, which should contain a series of
lines containing the reading name, position, and then an arbitrary
text comment. Once loaded a new dialogue allows stepping forwards and
backwards along the list. This is a replacement for the original
contig editor Search By File method.
Reading selection and Disassemble Readings
The Disassemble Readings function has been completely rewritten (along
with Break Contig too). It is now much much faster, but the key change
for functionality is the "Move readings to new contigs" and "Split
into single-read contigs" options. Moving readings to new contigs will
try to keep the assembly together as far as is possible. So for
example it is now possible to select all members of a repeat falsely
assembled and to disassemble the repeat to form one new contig (rather
than one per reading as in the previous Gap4, and as per the "split
into single-read contigs" option).
From this it follows that disassembling a reading and all readings to
its right is directly equivalent to the old break contig
function. Indeed Break Contig has now been written to do just this,
but it is still available as before.
This means that production of the lists of readings to pass into
disassemble readings has undergone a much needed improvement.
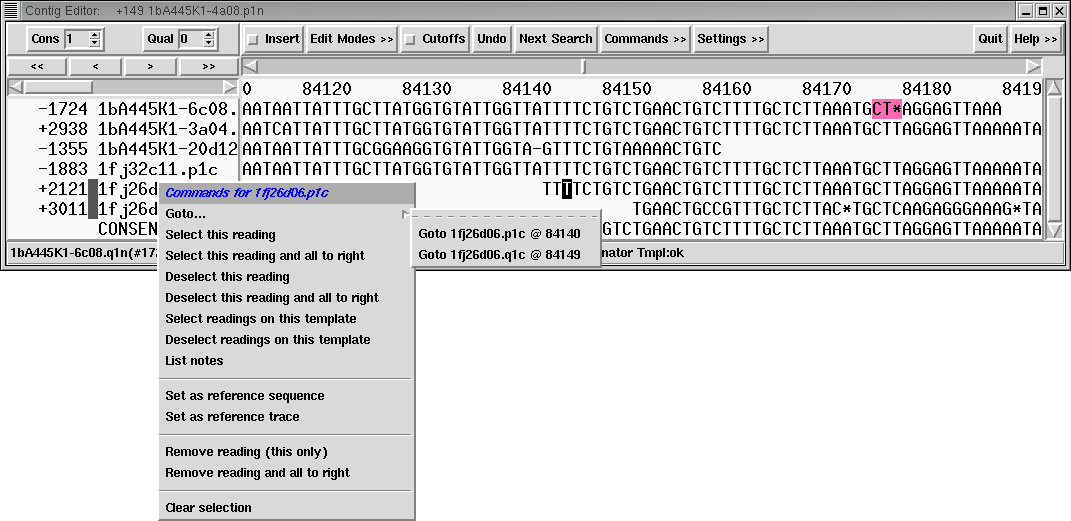
Manual generation of lists of readings from within the editor has been
simplified. There is no longer a difference between clicking using the
left mouse button and the middle mouse button. Additionally the popup
menu (via the right mouse button) on the reading names panel contains
options to "Select this reading and all to right" and "Deselect this
reading and all to right". (It is recommended that you use these while
in the editor "sort by positions" mode so that the consequences will
be obvious.)
Together these functions serve an easy way of selecting all readings
within designated regions. Furthermore, once selected the list can be
manually adjusted in the usual way by clicking on/off the highlight
for individual sequences.
Template information in the editor
The Contig Editor name panel has a coloured character (between the
reading number and name) to encode the consistency of the template. It
is white when consistent, so only inconsistent templates will have a
colour associated with them. The template consistency is also shown in
the status line of the editor when hovering the mouse over a reading
name.
The meanings are:
-
D / Light Grey
- Length of template ("D"istance). The size is
computed either from using the position of
forward and reverse readings or from the SVEC
vector tags on a single reading.
-
S / Red
- Strand. Either two forward (or two reverse)
sequences are on opposing strands, or a
forward and reverse read pair are on the same
strand.
-
P / Blue
- Primer position. If the start or end of the
template is computed using multiple sequences
(eg via two forward readings) and the start or
end is not consistently determined within
100bp then this is flagged.
-
? / Dark Grey
- More than one of the above inconsistencies.
When making a join in the join editor, using quit/join to make the
join will now inform you how many read-pairs span this contig, and how
many of those are consistent or inconsistent. (This information is
shown before you accept the join.)
- - -
And finally... the bionet/usenet newsgroup (bionet.software.staden)
seems very underutilised, but it can also be hard to post there
sometimes. The SourceForge forums are available at:
https://sourceforge.net/forum/?group_id=100316
If you wish to be kept up to date with new package releases and any
announcements, please either subscribe to the staden-package mailing
list held at sourceforge here:
https://sourceforge.net/mail/?group_id=100316
or create a SourceForge account and add a monitor to the 'staden'
package.
~ James Bonfield
Full Change List
Gap4
New features
- Added Contig Navigation to the main view menu. This is a GUI
to the GapNav (Sanger) tool, but equally well can take input
from any similar formatted file. (It is the same format that
Search by File in the editor uses.)
- Added Tag Editor "Move" and "Copy" buttons. To use these
underline the new tag location in the contig editor and then
hit the appropriate button in the tag editor. This may be used
to move tags to different sequences, the consensus, or even
different contigs.
- Tag Macros have been added to the contig editor. Pressing
Shift-F1 to Shift-10 brings up the macro editor. Then
underlining a region and pressing F1 to F10 generates the
appropriate tag type. (Your window manager may block some of
these keys, so experimentation will be called for.)
Tag Macros can also be generated by highlighting a tag and
pressing Control-F1 to Control-F10. This copies the tag
underneath the editing cursor to the appropriate macro number,
and so can be considered as a rudimentary cut and past.
- Fast editing and deletion of tags via the F11 (edit) and F12
(delete) keys. Note that F12 needs to be enabled in the Edit
Modes menu first.
- The Contig Editor names window now has a "goto..." submenu
allowing jumping to other readings from the same template
(whether or not they are in the same contig).
- Added a "Group by" submenu (of settings) to the Contig
Editor. This allows the reading names to be sorted by
position, name, template and strand.
- Rewrote disassemble readings. It is now much faster and is
also more flexible, allowing the marked readings to be moved
en-mass to new contigs while keeping their overlaps
intact. (Break contig is now just a special case of
disassemble readings, and has been rewritten accordingly.)
- Improved reading selection in the Contig Editor names
window. In addition to the usual left-click to highlight a
reading name, there is now a right-click menu containing a
variety of selection steps, such as all readings on this
template and all readings to the right of this point (useful
for a more fine-grained break contig via the disassemble
readings function). The 'set active list' output has been
improved too, so that this list now has "hyperlinks" when
viewed.
- Rewrote the Options->Set Fonts dialogue.
- Template status is shown in the editor name panel and the
status line.
- The count of consistent and inconsistent contig-spanning
templates is reported by the Join Editor before making the join.
- The Edit Tag command in the contig editor is now a cascading
menu listing all tags underneath the editing cursor. Similarly
for Delete Tag.
- The X11-based Tk file browser is now MUCH faster (between 80
and 160 fold).
- New command "N-base clip" in the main edit menu. This is to
work around a bug of phrap where it commonly adds long runs of
- or Ns just to include one extra base that, by chance, agrees
with the consensus.
- "Find sequence" (main view menu) now removes pads from the
query sequence before searching. This function also now allows
searching for matches within the reading sequences in addition
to within the contig sequence.
- The tables/*rc file loading now looks for *rc.local too. This
makes upgrading from one release to another easier.
- Save consensus now has the option to use the left-most
template name instead of left-most reading name as the contig
identifier. This is useful primarily for cDNA based projects.
- Reading numbers in the contig editor now have an explicit sign
(ie "+10" instead of "10").
- Added a GC_Clamp, self_any, self_end, max_poly_x and
max_end_stability options (from Primer3) to the oligo
selection dialogue and it now also remembers the users inputs
for other parts of this dialogue.
- The List Load function now automatically adds hyperlinks to
the input reading names meaning that it is a very useful way
of stepping through a list of sequences to
inspect/edit. (However see Contig Navigation for an ever
better way.)
- add_tags and enter_tags API functions now allow for tags to be
loaded with unpadded sequence coordinates.
- Improved output to the database .log file. It now contains
user name and hostnames.
- The contig identier component of dialogues now automatically
has focus and selection, allowing for quicker overtyping of
the contig name.
- Right-clicking on a tag in the contig selector now allows for
"edit contig at this tag".
- Remember the editor Search window values (within a single
session) to speed up searching for the same thing over
multiple contigs.
- Check Database now checks that all sequences contain printable
characters. This slows it down by approximately 25%.
- The RAWDATA search path now allows for traces to be fetched
directly from the ensembl trace repository or via any
specified URL (via the wget utility).
- The List Contigs window now has a save button to save the
contig order (useful after sorting by column headings).
Bug fixes
- The consensus algorithm was treating bases in columns with
pads a little differently than bases in columns without
pads. This has now been sanitised to be consistent, although
it may yield confidence values 1 or 2 lower than before in
some cases.
- Improved error reporting from disassemble readings and break
contig.
- Improved error reporting of tag input.
- Various template calculations (consistency, coordinates) have
been bug-fixed. This is primarily a fix for prefinish, but the
bug was in gap4 and this also improves other parts of gap4
too. Also the orientation for inconsistency templates is now
set to the most likely orientation rather than "?".
- The trace display should now load faster due to removal of the
file format checking (which caused every trace to be loaded twice).
- Find Internal Joins was missing some matches. Additionally
there are tighter constraints on it uses up lots of memory
and/or CPU time. It also depads sequences prior to searching
for alignments, which helps with very deep alignments.
- Fixed a problem causing database files to gradually grow
unnecessarily.
- Sped up the output for extract_readings when saving in
directed assembly format.
- Restriction enzyme map: corrected the textual output to count
in unpadded base coordinates (was padded).
- Removed a buffer overflow in the tag-drawing code of the
contig selector.
- Fixed generation of the mutation report when the assembly
contained sequences without traces or with missing traces.
- Tag searches (and maybe others) in the editor could miss hits
when Group Readings by Templates was enabled.
- The "Select All" and "Clear All" buttons in the tag selector
windows are back. (They were removed by accident.)
Prefinish
Changes/New features
- Apply a cost increase to experiments that do not connect
to at least one end of the problem region.
The reason being that a problem from 1-1000 with reading length 600
can be solved in 2 experiments, but chosing the first experiment
(due to minor reasons like better primer) from 200-800 requires 2
extra experiments to solve it.
- Added a two-pass method to generate_experiments. Pick
the most appropriate end for single-stranded experiments (as
before), but if that fails we now work outwards from the other end
too. This helps to pick experiments when our contig is covered
entirely by a single-stranded region.
- New option -skip_fake_templates to reject templates that do
not contain at least one reading referring to a trace
file. This is designed to be an easy way of filtering out
assembled consensus sequences.
- Added a notion of a result type having a desired number of
solutions to use. This is distinct from the number of items in a
group. Eg we may want to pick 2 primers, and use 3 templates for
each. The main purpose for this though is for picking more than 1
resequence experiment for each problem.
Added -reseq_nsolutions, -long_nsolutions and -pwalk_nsolutions
as configuration parameters.
- Improved the score of experiments that have large groups. Eg more
templates for a primer-walk are better than one.
- Added the filter_words algorithm into prefinish so that poly-A,
GT-rich, etc can be filtered on. These are now also defined
classification types and we check for different characters other
than '#' in the finish_walk algorithms too.
- Added a bonus score for experiments that fix all mandatory
problems within a +/- 100 base pair region. This gives
an encouragement to not leave tiny problems which require another
experiment.
Bug fixes
- Template start and end coordinates for inconsistent templates
are not computed via the min/max observed locations rather
instead of the computed ranges, as the computation is often
wrong with inconsistent data.
- Fixed a bug in the use of strand vs sense. We now check sense for
resequencing experiments as it doesn't matter if it's fwd or
reverse sequence, just that it heads in the right direction.
- Speed up by moving the dust filtering to before primer picking
instead of after. This removes the generation of lots of false
primers.
- Added code to correctly score sequences that extend the contig (if
extending is required) even though the problem being solved may
not be the contig-extend problem.
In this case it also correct clears the CONTIG_LEFT_END and
CONTIG_RIGHT_END classification flags.
Mutscan
Changes/New features
- Filters clusters of tags as these are invariably due to
alignment failures.
Bug fixes
- Correct for divide by zero and log(0) in various places.
Io_lib
- Added LG (Ligation - a combination of LI and LE) to the
experiment file format.
- Added a -fofn option to extract_seq.
- Better error checking for writing compressed files.
- Protect against the base spacing being listed as a negative number
in the ABI file.
- Added support for reading phred-style confidence values from
ABI files.
- io_lib can now fetch traces directly via a URL or from the
ensembl trace repository using either URL=%s or ARC=%s:%d
(host:port) syntax.
Misc
- New program "stops". It searches for likely 'stop' regions
within trace files.